






翻译后修饰(PTM)是调控蛋白质功能与相互作用的关键机制,其异常与疾病相关。错义变异可通过破坏、新增或影响邻近PTM位点改变修饰模式。现有预测工具如MIMP局限于特定激酶或磷酸化,而新兴深度学习模型虽扩展了预测范围,却因依赖未经统一质控的公共数据库(如PhosphoSitePlus)导致准确性受限。

作者通过系统性研究构建了PTMAtlas数据库并开发了DeepMVP预测模型(图1)。首先通过对公共质谱数据集进行严格质量控制,生成了包含高置信度PTM位点的PTMAtlas。基于此,作者开发了酶非特异性的深度学习模型DeepMVP,其采用经遗传算法优化的CNN-BiGRU混合架构与集成学习策略,可预测六类PTM位点。为评估变异效应,DeepMVP通过计算参考序列与变异序列的PTM概率差异生成差分分数(正/负值分别表示PTM可能性增加/降低),该分数可识别变异位置(直接影响)或邻近位点(近端效应)的PTM改变。PTMAtlas与DeepMVP均已公开,为PTM功能研究提供资源与工具支持。
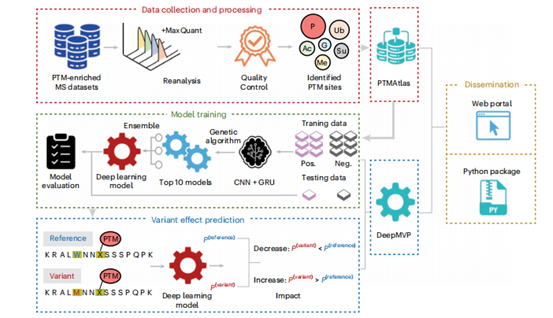
图1 | 研究设计概览
PTMAtlas:一个全面、高质量的PTM位
点数据库
作者通过对241个人类PTM富集MS/MS数据集(含20,675个原始文件)进行系统性重分析(图2a),构建了包含397,524个高置信度PTM位点的PTMAtlas数据库。该数据库涵盖六类修饰:乙酰化(33,010位点)、甲基化(15,843位点)、N-糖基化(7,920位点)、磷酸化(194,903位点)、SUMO化(39,071位点)和泛素化(106,777位点)(图2a)。通过严格FDR控制(1%)和定位概率筛选(>0.5),PTMAtlas展现出显著的质量优势:其磷酸化位点数量是UniProt的5倍,且新增83,162个PSP未收录位点(图2h);在乙酰化、甲基化等修饰类型中位点数量全面领先现有数据库(图2e-j)。稀疏曲线分析显示除N-糖基化外多数PTM类型尚未饱和(图2b),而与其他数据库的对比验证了PTMAtlas在覆盖广度与数据质量方面的综合优势。
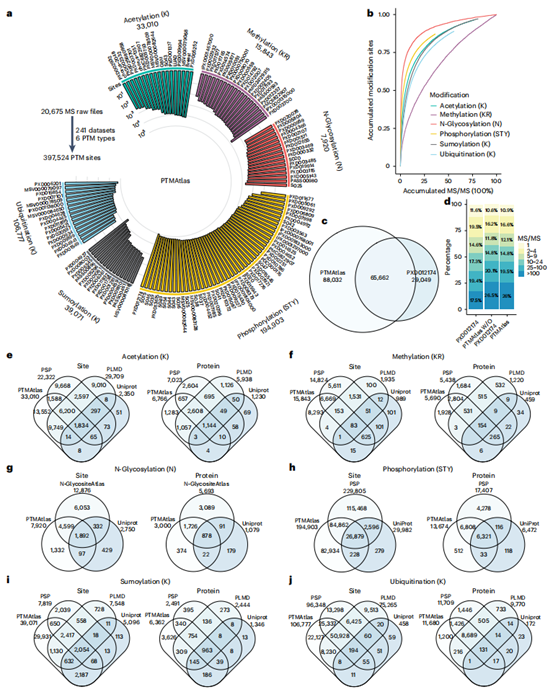
图2 | PTMAtlas的创建与其他PTM数据库的比较
基于深度学习的 PTM 位点预测
作者基于PTMAtlas数据库开发了DeepMVP深度学习模型(扩展数据图3a-c),该模型在六种PTM类型(磷酸化、乙酰化等)的位点预测中全面超越现有工具(图3a-h),所有PTM类型的AUROC均高于0.85(磷酸化达0.95)。严格的序列相似性控制验证表明其性能稳定,且模型优势源于神经网络架构优化与高质量训练数据的结合。DeepMVP实现了全蛋白质组规模预测(图3i),覆盖99%以上人类蛋白质及SARS-CoV-2病毒蛋白,并成功验证了病毒蛋白中80%-87.5%的实验鉴定磷酸化位点(图3j)。这些结果证实DeepMVP通过整合先进算法与高质量数据,显著提升了PTM位点预测的准确性与泛化能力。
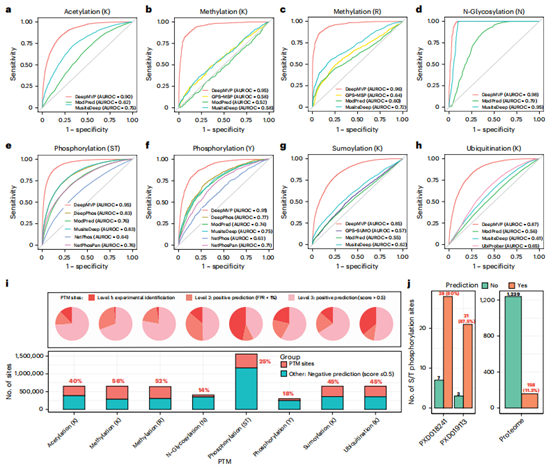
图3 | DeepMVP在PTM位点预测中的评估与应用
预测变异对PTM的影响
作者通过文献整理的235对实验验证变异-PTM组合(图4a)和CPTAC癌症队列(子宫内膜癌与肺鳞癌)的蛋白质组学数据(图4c-d)评估了DeepMVP预测变异对PTM影响的能力。DeepMVP在文献数据中表现出高精度:对PTM位点存在的预测准确率达81%(191/235),对变异影响方向的预测整体准确率为97%(直接事件达100%)(图4b)。在癌症队列分析中,DeepMVP识别出3,365个PTM改变事件,其中98%(3,296个)获MS/MS数据支持,冲突率仅2%(图4d),其敏感性和特异性均优于MusiteDeep、MIMP等工具(图4c)。这些结果证实DeepMVP能可靠预测变异对PTM的直接与邻近效应,且与实验数据高度一致。
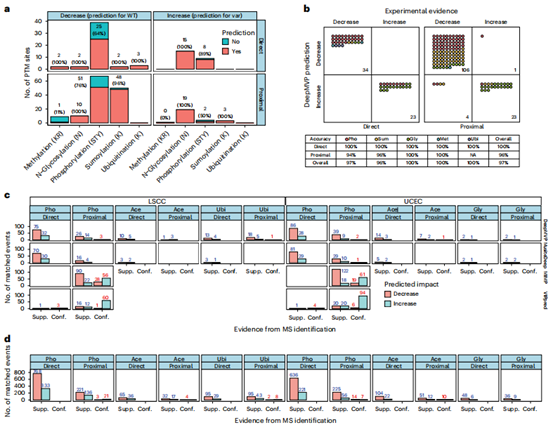
图4 | PTM变异和蛋白质基因组学数据集验证变异效应预测
疾病相关生殖系变异的PTM效应预测
作者利用DeepMVP结合Shapley值分析(图5b,d,f)系统评估了ClinVar中24,237个致病性变异对PTM的影响(图5a),预测出7,713个变异(32%)显著改变12,435个PTM事件。该模型准确捕获了已知病理机制:如LMNA S22A变异消除磷酸化导致心脏传导疾病(扩展数据图6a)、RAF1 S259F变异激活致癌信号(扩展数据图6b),以及FBN1 I1048T变异引入N-糖基化致马凡综合征(图5b)。同时,DeepMVP揭示了未报道的致病机制:如SCNN1B P616L变异通过破坏CDK激酶基序减少T615磷酸化(图5e-f),可能导致高血压;FUS R524S变异通过增强Y526磷酸化(图5g)促进ALS病理。这些预测不仅通过Shapley值分析提供了可解释的分子机制(如基序破坏/形成),还为靶向修饰酶的治疗策略提供了新方向。
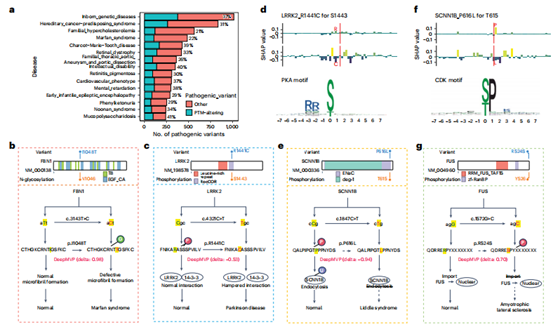
图5 | DeepMVP对PTM改变致病性生殖系变异的预测
泛癌分析体细胞突变对PTM的影响
作者利用DeepMVP分析了33种癌症类型中9,079个样本的791,637个体细胞突变(图6a),预测31%(230,092个)的突变会改变PTM状态。该模型揭示了新的致癌机制:如AKT1 E17K通过减少K20乙酰化(抑制性修饰)驱动激活(图6b);TP53 G266R通过形成RRXS/T基序增加S269磷酸化(图6c-d),实验验证显示突变型中S269磷酸化特异性出现(图6g);VHL L169P通过形成S/TP基序促进S168磷酸化(图6e-f),HeLa细胞实验证实突变特异性磷酸化(图6g)。这些发现将未知功能突变与PTM失调联系起来,并通过Shapley值分析揭示了潜在激酶机制。DeepMVP已集成至网络服务器与Python包,支持广泛研究应用。
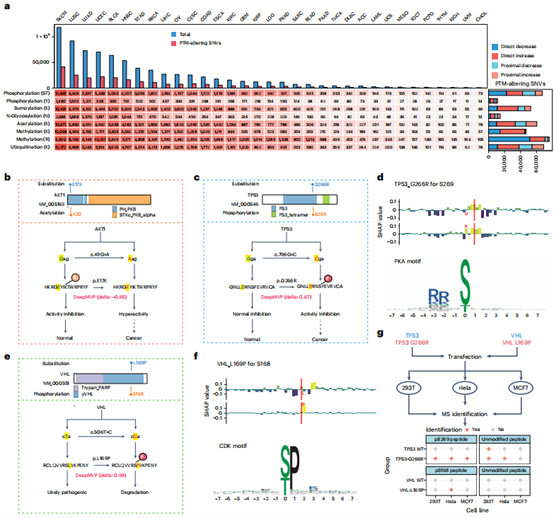
图6 | DeepMVP对33种癌症类型中PTM改变体细胞突变的预测
总结
DeepMVP通过整合CNN-GRU混合架构与遗传算法优化(图6相关),利用高质量PTMAtlas训练数据,实现了对六种PTM类型变异效应的全蛋白质组预测,成功识别出7,713个致病性生殖系变异和230,092个癌症体细胞突变中的PTM改变事件。其酶非特异性设计结合Shapley值可解释性分析,可推断潜在修饰酶机制(如AKT1 E17K减少K20乙酰化、TP53 G266R形成激酶基序)。虽存在胰蛋白酶偏倚和结构上下文建模局限,但经文献与癌症蛋白质组学数据验证(图4,6g),DeepMVP显著优于现有工具,为变异-表型关联提供了PTM层面的功能解析,填补了传统方法无法揭示特异性分子机制的空白。

