






在生命的浩瀚反应网络中,酶就像无数个“分子工匠”。它们决定了细胞中成千上万种化学反应的发生与否,也塑造了生命系统的复杂性与精准性。然而,人类对酶的理解仍然有限。尽管我们已经测定了数百万条酶序列,但对于其中大部分,我们仍然无法回答一个最核心的问题:“这条酶到底能识别哪种底物?”
这正是结构生物学与人工智能交汇的前沿挑战。传统的实验筛选和序列比对往往效率低、依赖模板、缺乏泛化能力。而最近,《Nature》发表的一项研究让人眼前一亮——来自伊利诺伊大学厄本那–香槟分校(UIUC)Huimin Zhao 和 Diwakar Shukla 团队的最新成果,提出了一种能够“读懂酶语言”的人工智能模型 EZSpecificity,它利用深度学习真正理解了酶与底物之间的化学识别逻辑。

这项研究的突破首先来自于一个前所未有的大型数据库。团队建立了名为 ESIbank(Enzyme–Substrate Interaction Bank) 的资源库,它整合了来自 BRENDA、UniProt、AlphaFold 等多个数据库的信息,并利用 GPU 加速的 AutoDock 算法对每个酶–底物组合进行了结构对接。最终,他们得到了一个包含 8,000 多种酶、34,000 多种底物、超过 32 万条酶–底物关系 的三维数据集——这几乎是此前公开资源规模的 25 倍。这为人工智能模型提供了丰富的“生物语言语料库”。EZSpecificity 正是在这样的数据土壤上成长起来的。
作者如何从文献与公共数据库中整合出一个可用于训练 EZSpecificity 模型的大规模酶–底物相互作用数据集。整个流程分为数据来源-收集-数据清理与比对-对接与复合物生成-Database Integration → ESIbank (图1)
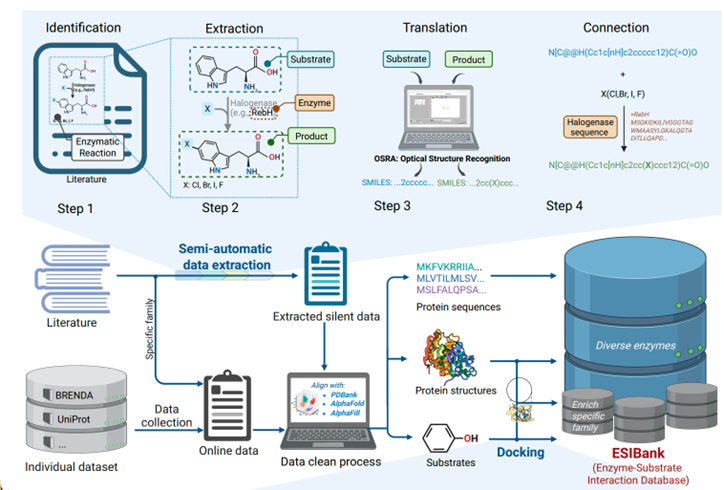
在数据收集这一步,作者发现,许多酶-底物信息并没有被收录进现有数据库,而是“藏”在论文图表里。所以他们设计了一个 半自动化的四步流程:
Step 1 – Identification 识别
从文献中识别酶催化反应图(例如卤化反应图)。自动检测哪些反应是由某类酶(如 Halogenase)催化的。
Step 2 – Extraction 提取
自动分离反应图中的 底物(substrate)、酶(enzyme) 与 产物(product)。图中举例:底物芳香环被卤化酶 Rebh 作用,生成带 Cl/Br/I/F 取代基的产物。
Step 3 – Translation 转换
使用 OSRA (Optical Structure Recognition Application) 将图像里的化学结构识别成机器可读的 SMILES 字符串。这样,模型就能直接理解底物与产物的化学信息。
Step 4 – Connection 关联
将酶序列与底物的 SMILES 式配对,生成酶-底物反应记录。图中展示了 Halogenase 序列与底物-产物 SMILES 的对应连接。
这四步结合识别与文本解析,实现了从图像中提取反应信息的自动化,大大扩展了可用数据量。
接着,作者将这些从文献获得的信息,与各类公开数据库结合:
Data Collection
从 BRENDA、UniProt 等数据库提取酶序列与功能信息;将文献抽取结果加入对应家族(如 Halogenase、Thiolase 等)。
Data Cleaning & Alignment
将酶序列与结构数据库(PDBank、AlphaFold、AlphaFill)对齐;检查缺失或异常数据。
Docking Process
对每个酶–底物对进行 AutoDock GPU 对接,生成复合物结构;这些结构提供了模型训练所需的三维结合位点信息。
Final Integration → ESIbank
最终生成的数据库包含:酶序列(Protein sequences)酶结构(Protein structures)底物 SMILES 与对接姿势(Substrates + Docking)
在拥有数据集之后,作者开始搭建EZSpecificity 模型,那如何从序列 + 结构 + 底物三类信息中,学习酶的底物特异性。可以把它理解成一个“三步走”的系统(图2):
左边:准备输入(蛋白序列、底物分子、结构)
中间:两种编码(序列编码 + 活性口袋编码)
右边:交叉注意力融合 → 输出预测特异性
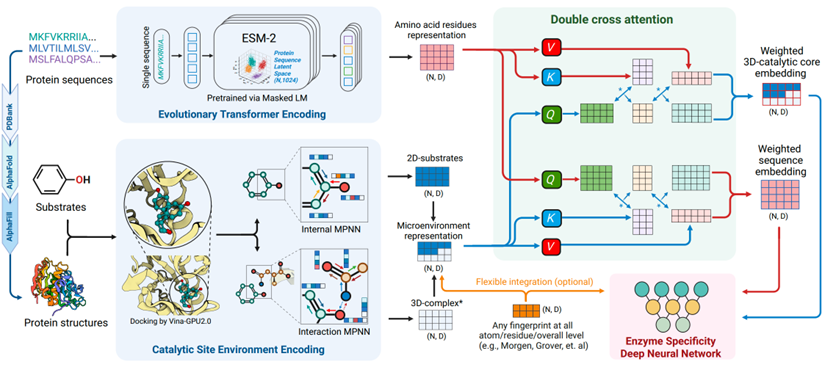
什么是 ESM-2?
ESM-2(Evolutionary Scale Modeling 2)是 Meta AI 团队开发的一个 蛋白语言模型(Protein Language Model, PLM),相当于生物序列界的 GPT。在 ESM-2 中,模型会随机遮盖序列中的部分氨基酸(例如 15% 的位置),然后让模型根据上下文去“预测”被遮盖的残基是什么。这意味着模型在学习“一个氨基酸在整个序列上下文中的语义角色”,也就是:某个位置出现某个残基的概率取决于其他残基的组合。久而久之,模型就会自发学习出类似:
哪些氨基酸常在 α-helix 区域;
哪些序列模式出现在活性位点;
哪些远程相互作用模式对应折叠拓扑。
换句话说,它在“语言层面”学会了结构和功能规律。
在经过 ESM-2 编码后,每个氨基酸会被映射到一个高维向量空间(通常是 1,280 维),形成一个 N × D 的矩阵:
N = 残基数
D = 向量维度
这就相当于为每个氨基酸建立了一份“语义档案”,
其中包含:
它的进化背景(通过大量序列共现模式学习得来);
它在结构中的可能角色(如亲水/疏水倾向);
它对底物识别的潜在贡献。
这些特征随后被送入交叉注意力模块,与底物分子的特征结合。
Catalytic Site Environment Encoding(活性位点环境编码),这部分捕捉三维结构信息:对每个酶–底物复合物,模型会关注活性口袋周围的原子。使用 SE(3)-equivariant Graph Neural Network (GNN) 进行建模。SE(3) 意味着模型对旋转和平移不敏感。每个节点代表一个原子(来自酶或底物),边代表化学键或空间邻近关系。
图中展示了两种 MPNN(消息传递神经网络):
Internal MPNN:在分子内部传递信息(捕捉局部结构)。
Interaction MPNN:在酶和底物之间传递信息(捕捉结合位点相互作用)。
最终得到蓝色矩阵 “Microenvironment representation”,描述了原子级别的局部环境。
“双重”交叉注意力的创新点:
作者并没有只让酶关注底物,而是双向关注。
酶残基作为 Query,底物原子作为 Key/Value,得到“哪些残基最关注哪些底物”。
这对应我们理解的活性位点识别模式。
底物原子作为 Query,酶残基作为 Key/Value,得到“哪些底物原子最依赖哪些酶环境”。
双向交叉注意力相当于让模型从两个角度去理解结合界面。
经过双向注意力后,模型得到了两个核心结果:
Weighted 3D Catalytic Core Embedding(加权三维催化核心表示)
→ 强调那些在结合界面上贡献最大的结构原子(比如口袋中的关键残基)。
Weighted Sequence Embedding(加权序列表示)
→ 强调那些在进化上保守且功能上重要的序列片段。
这两个结果会被合并输入下游的深度神经网络,用来预测酶–底物的匹配分数。
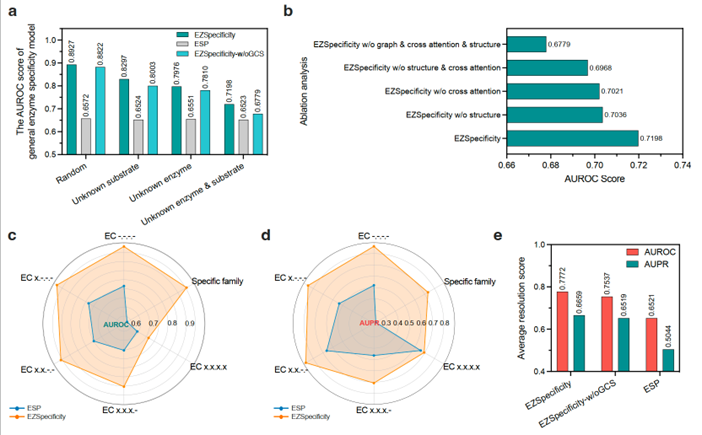
接下来,作者系统评估了 EZSpecificity 模型的表现,包括五个部分(图3):
(a) 模型在不同测试集下的总体性能;
(b) 消融实验,测试各个模块的重要性;
(c–d) 在不同 EC 分类层级下的表现;
(e) 模型的平均预测分辨率。
通过这些结果,作者证明 EZSpecificity 在泛化性(unknown enzyme/substrate)和模块有效性上都有显著提升。
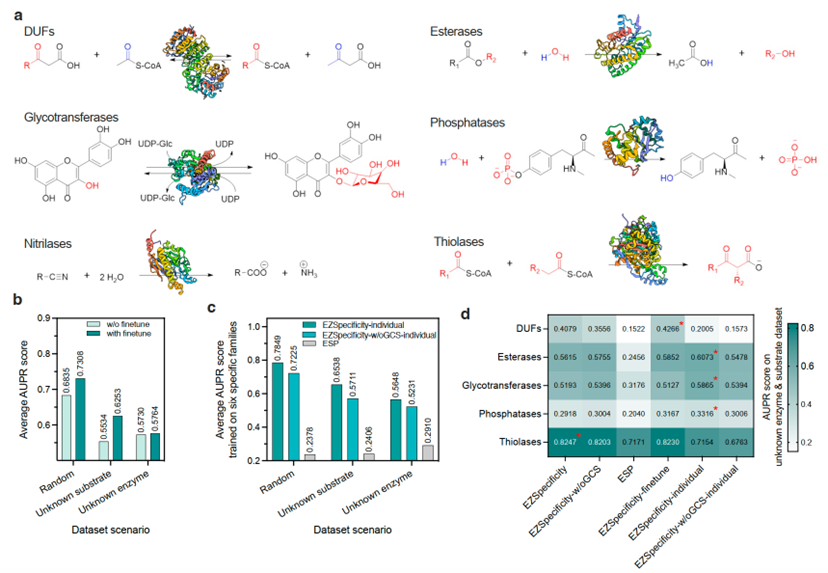
验证模型:展示了 EZSpecificity 模型在不同酶家族中的泛化表现,也就是它能否在真实的酶系统中跨家族预测底物特异性。作者选了六类典型的家族,它们在结构、底物类型和反应机理上差异极大,因此非常适合测试模型的跨家族泛化能力(图4)。
作者评估了在不同数据场景下,模型是否需要进行微调(fine-tuning)。
浅蓝色柱:未微调模型(w/o finetune)
深蓝色柱:微调后模型(with finetune)在原始的 EZSpecificity 模型基础上,利用某个目标酶家族的数据(例如 esterase、glycosyltransferase 等)继续训练若干个迭代。
总结
这个模型有三个非常创新的点。
第一,它引入了 cross-attention(交叉注意力)机制。
什么意思呢?模型会自动学习“哪一个氨基酸残基”和“底物的哪一个原子”最相关,这就像让 AI 自己去“看”哪里在起关键作用。
第二,它使用 SE(3)-equivariant GNN。
这是一种可以理解三维结构的图神经网络,不论分子怎么旋转或平移,模型的判断都不变。
第三,它把 ESM-2 序列模型、AutoDock 生成的结构、和分子图特征三者结合,真正把“序列—结构—相互作用”统一到一个体系中。
特征与 docking 结构,从多个层面理解酶–底物识别。”
当然,模型也有局限性。
目前它只能预测底物特异性,还不能区分反应的区域选择性(regioselectivity)或立体选择性(stereoselectivity)。
而且模型依赖 docking 生成的结构,如果 docking pose 不准,也会带来误差。
此外,不同 EC 分类的数据量仍不均衡,这可能造成部分酶家族的泛化能力不足。
作者还提到,下一步将加入 分子动力学(MD)和量化化学(QM)信息,进一步理解反应的动态特征。

