






生物催化因其条件温和、催化选择性高以及绿色环保等优点,正日益成为合成化学、制药、精细化学乃至材料科学中的重要工具。然而,尽管酶催化具备这些显著优势,其在新底物、新反应通路中的广泛应用仍然面临重大瓶颈。首先,已知酶-底物配对的数据极其有限:大部分酶的底物谱未知,大量可催化潜力尚未被挖掘。其次,传统酶催化探索往往依赖大规模实验筛选、酶突变优化或工程进化,这不仅耗时耗力,而且实际成功率较低。从化学空间到蛋白序列空间之间,缺乏足够连接“哪个酶能催化哪个底物”的映射,使得科研人员在实际合成规划中往往需要盲目尝试、成本高风险大。在这样的背景下,Alison R. H. Narayan研究团队提出了一个核心动机:构建一个高质量、广覆盖的酶-底物连接数据库,并利用机器学习模型将化学空间与蛋白序列空间“连接”起来,从而实现从底物到酶、或酶到底物的预测推荐。换句话说,他们希望从“我有这个化合物,哪个酶可以催化?”或“我有这个酶,它可能催化哪些尚未尝试的底物?”的角度,为生物催化领域提供一种系统化、可扩展、数据驱动的新范式。
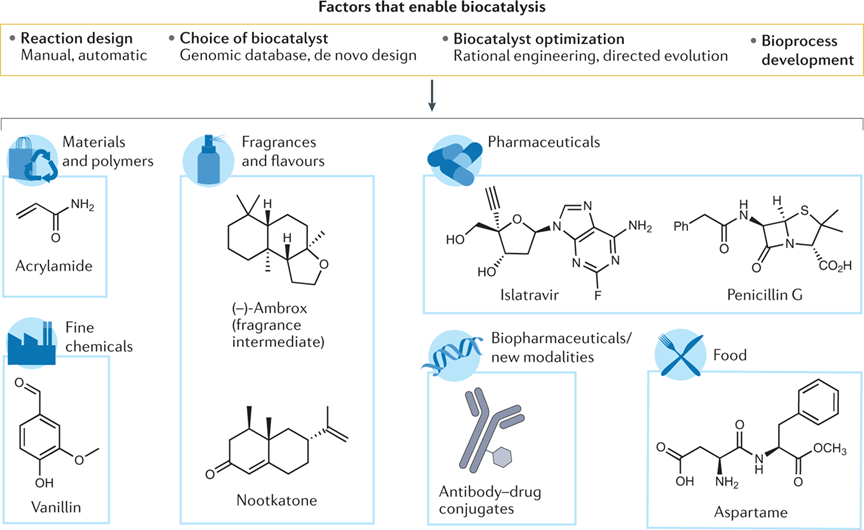
生物催化的重要性
2025年10月1日,美国密歇根大学Alison R. H. Narayan和卡耐基梅隆大学Gabe Gomes团队合作在顶级期刊《Nature》上发表了一篇题为“Connecting chemical and protein sequence space to predict biocatalytic reactions”的研究,系统介绍其设计思路、数据构建、模型开发与应用前景。研究团队首先构建了大规模酶-底物反应数据库,覆盖数百位酶与上百种底物,从而填补此前 “酶-底物配对稀疏” 的空白。继而,基于该数据库开发了推荐型机器学习平台 CATNIP,使得给定底物可预测适配酶、或给定酶可预测潜在底物。通过该研究可见,生物催化已迈入 “从经验筛选转向数据驱动推荐” 的新阶段,对催化剂发现、合成路线设计、绿色化学发展具有重要意义。

高通量实验筛选
研究团队选择了一组 α-酮戊二酸(α-KG)依赖非血红素铁(NHI)酶作为研究对象,构建酶库,并对底物库中的超过 100 种分子进行反应测试,以生成大量酶-底物配对数据。该过程通过高通量自动化平台、96孔板或更高通量微孔体系、统一反应条件、标准化分析(如 LC-MS)进行,从而为数据驱动分析奠定基础。此阶段的目的不仅是生成阳性反应(酶催化成功)数据,还包括无反应/弱反应的负样本,为后续模型训练提供更全面的数据基础。
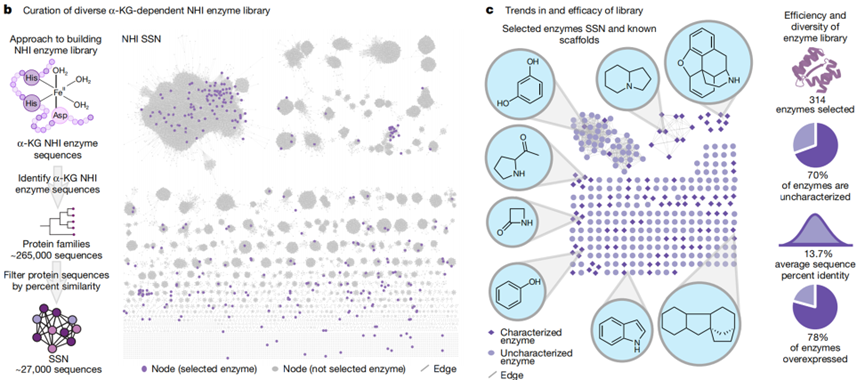
图1. α-酮戊二酸非血红素铁酶多样性的原理及 aKGLib1 库的构建
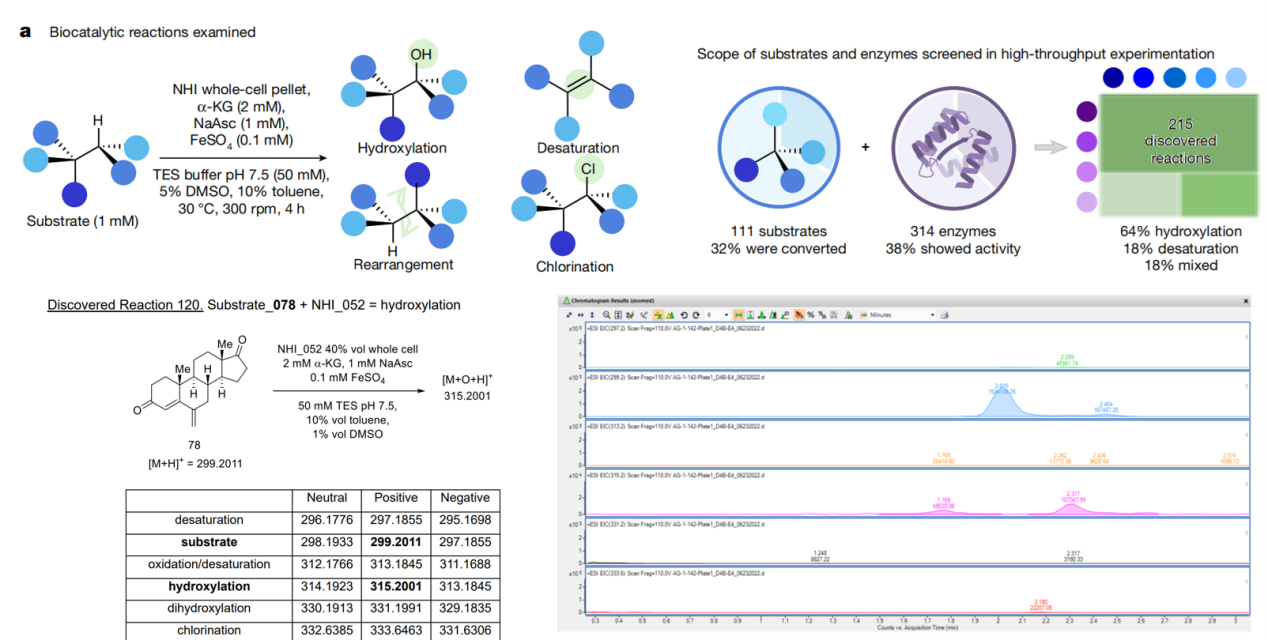
图2. 高通量反应发现工作流程
数据集与特征工程
在数据集构建方面,酶库主要聚焦 α-KG/Fe(II) 依赖的 NHI 酶家族,这类酶在 C–H 功能化)方面已具一定基础。研究者首先通过序列相似性网络(SSN)方法对酶序列进行聚类,选取代表序列以覆盖序列空间,从而保证酶库不仅限于少数已知活性酶,而扩展至未表征酶。底物库则选择结构多样的分子群,包括天然产物、化学合成建筑块、药物中间体、衍生分子等,以扩展化学空间。特征工程方面,酶序列特征包括对齐分数、序列身份、簇类别、保守区域指标等;底物特征则包括传统2D指纹、分子量、LogP、极性/疏水性指标、以及3D几何描述符。此外,模型还可能考虑酶-底物邻居关系作为迁移推荐依据。最终,训练数据中每对酶-底物组合包含标签,并为模型提供正/负样本。通过这种系统设计,研究团队实现特征维度多样且覆盖广泛,使得模型具备更强泛化能力。
机器学习模型构建与应用
基于阶段一获得的酶-底物配对数据库(BioCatSet1),研究者提取酶序列特征底物化学/几何特征,并构建推荐型机器学习模型,名为 CATNIP。模型的核心思想是:给定一个底物,输出一个酶排序;或给定一个酶,输出其潜在底物排序。特征工程包括酶的序列相似性网络 (SSN)、序列对齐分数、酶与酶之间的距离、底物的分子指纹、3D几何描述(如 MORFEUS 描述符)等。模型训练使用交叉验证、分割训练/验证/测试数据集,并通过 Precision@k、Recall、nDCG(归一化累积折损增益)等指标评估性能。报告中还明确指出,他们将该工具以开放网页形式(CATNIP Web界面)提供给社区用。
通过这种思路,研究团队不仅生成了丰富的数据资源,还通过模型构建完成“化学空间到蛋白序列空间”的链接,使得生物催化的酶选择从传统“经验+筛选”转变为“数据驱动荐”。
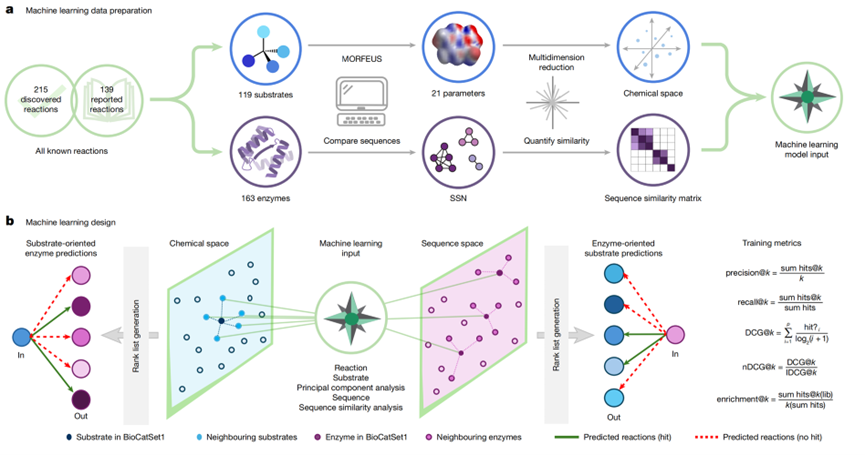
图3. 从反应数据到机器学习模型的转换
模型构建与性能评估
对于模型构建,研究团队将获得的数据分为训练集、验证集与测试集,确保评估公平。模型采用推荐系统思路:对于底物 → 酶任务,模型输出酶排序;对于酶 → 底物任务亦然。评价指标包括 Precision@k(前 k 项中真实活性配对所占比例)、Recall(识别所有活性配对的比例)、nDCG(考虑排序和位置权重的指标)等。模型的基线包含传统距离/相似性排序模型(如最近邻推荐、仅化学指纹匹配、仅序列相似排序)等。结果表明:该模型在多个任务情况下优于这些基线,显示出更高的前 k 推荐准确率,并且在“酶从未用过底物”或“底物从未用过酶”这类挑战性任务上仍有可用性能。此外,研究团队还探讨了模型可解释性:例如通过分析酶-底物邻域迁移(即底物若与某已知底物相似,则其推荐酶可能为该底物对应酶的邻近酶)、以及特征贡献分析(如底物几何特征在预测中权重较高)等。这种结合实验、特征工程、机器学习推荐、应用网页工具的流程,使得模型不仅是算法演示,更具实用性和可操作性。
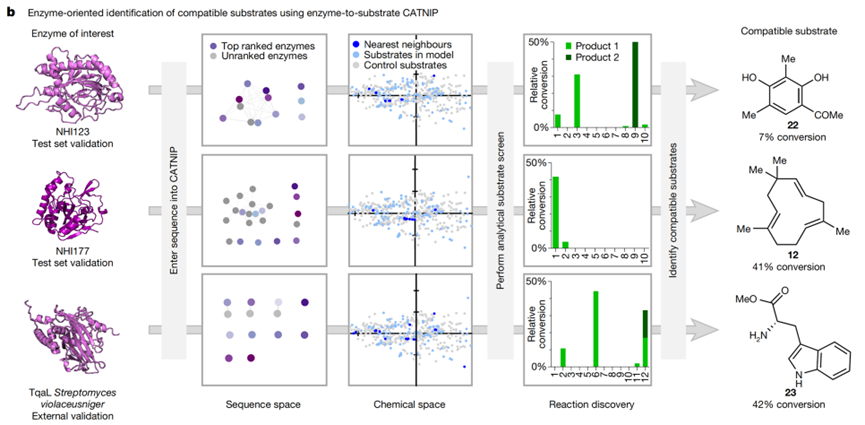
图4. CATNIP:一款用于预测生物催化反应的网络应用程序
本研究具有广泛而深远的应用意义。首先,在催化剂发现方面,化学家可借助 CATNIP 平台,在合成路线规划阶段快速筛选可能适配的酶,大幅缩短酶筛选与优化周期。其次,在绿色合成领域,通过更快、准确地找到生物催化路径,有望降低化学合成中对重金属催化剂、有机溶剂或高温高压条件的依赖,从而提升反应的环境友好性和可持续性。再次,在新反应开发方面,该方法有助于 “new-to-nature” 的酶催化转化探索,即化学家可输入尚未用生物催化的底物,模型推荐酶,验证后可能开辟新的催化类型或功能化手段。最后,作为工具平台,CATNIP 的开放访问意味着社区研究者可直接使用、扩展至更多酶家族与反应类型,从而推动整体生物催化领域发展。

