






代谢稳态是维持细胞和机体功能的基础,其失衡广泛存在于感染、炎症性疾病和肿瘤等病理状态中。先天免疫信号的激活常与代谢通路紊乱并存,例如糖酵解或氧化磷酸化受损可诱导炎症反应和细胞死亡。此类代谢应激通常伴随线粒体功能障碍和氧化应激,从而推动炎症放大和组织损伤。尽管氧化应激已被证明参与多种受调控的细胞死亡形式,其如何在先天免疫激活与代谢紊乱协同作用下破坏细胞膜完整性并触发炎症性细胞死亡,仍缺乏清晰的机制解释。

在病理条件下,先天免疫激活常与营养限制并存,从而导致代谢紊乱。研究人员发现,在 LPS 诱导的脓毒性休克模型中,预禁食显著提高小鼠死亡率并加重组织损伤。在体外,研究人员在碳源匮乏(carbonstarvation,CS)条件下给予骨髓来源巨噬细胞(BMDMs)先天免疫刺激,以模拟疾病状态中先天免疫激活与代谢紊乱(IIAMD)的协同作用。结果显示IIAMD可在骨髓来源巨噬细胞中诱导显著的裂解性细胞死亡,而单独处理均不足以致死(图1)。该细胞死亡依赖代谢紊乱,补充葡萄糖、谷氨酰胺或丙酮酸可有效抑制其发生。IIAMD同时诱导 NLRP3 炎症小体激活及 IL-1β、IL-18 等炎症因子释放。该过程主要依赖 TLR–MyD88 信号轴,而非完全依赖 TNF 信号。
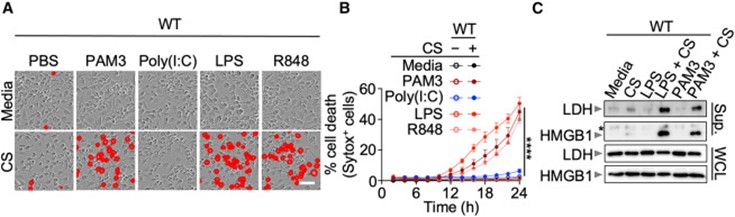
图1. IIAMD 协同诱导细胞死亡
NLRP3 通常感知稳态紊乱并诱导焦亡或 PANoptosis,然而 IIAMD 不依赖 NLRP3 及 gasdermin 家族或 MLKL 等已知成孔执行分子。尽管可检测到 caspase 激活和 MLKL 磷酸化,但抑制或遗传敲除凋亡、焦亡、坏死性凋亡、PANoptosis 或铁死亡相关关键分子均无法阻止细胞死亡。NINJ1 对质膜破裂和 DAMPs 释放是必需的,但并非细胞死亡本身的执行因子。基于此,研究人员推测IIAMD 诱导的细胞死亡独立于凋亡、焦亡、坏死性凋亡、PANoptosis 和铁死亡等已知通路。
非靶向代谢组学分析显示,IIAMD 显著重塑细胞代谢谱,其中谷胱甘肽(GSH)代谢明显受损。GSH 耗竭导致氧化应激在细胞膜破裂和细胞死亡前逐渐累积,补充 GSH 或使用抗氧化剂 NAC 可有效抑制 IIAMD 诱导的细胞死亡(图2)。这一结果说明这些结果表明,IIAMD 通过耗竭 GSH 引发氧化应激,并经由一种此前未被描述的通路诱导细胞死亡。
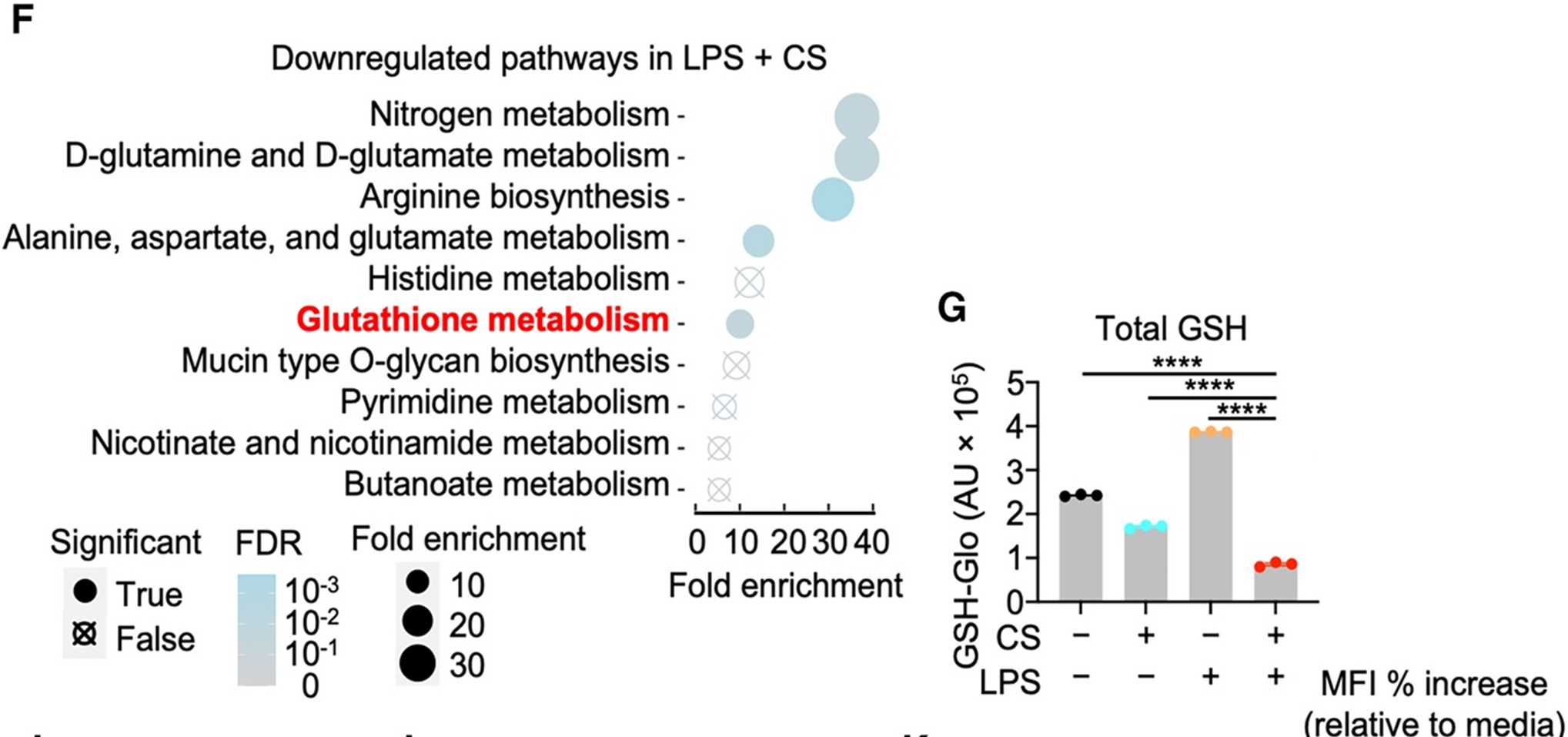
图2. IIAMD 通过耗竭 GSH 引发氧化应激
为进一步理解发生氧化应激的细胞器亚细胞动态与 IIAMD 诱导细胞死亡之间的关系,研究人员进行了共聚焦活细胞成像分析。结果显示在 IIAMD 条件下,发生氧化应激的细胞器长期定位于质膜附近,并在接触位点诱发膜退化和最终破裂。共定位分析和定量分析证实IIAMD可以显著延长了线粒体–质膜的结合时间,该持续性线粒体–质膜接触是 IIAMD 诱导细胞死亡的特异性特征。
鉴于活性氧主要由线粒体产生且作用范围有限,研究人员推测 IIAMD 诱导的持续线粒体–质膜接触会导致局部膜氧化损伤并触发细胞裂解。实验结果也证明了这一假设的正确性:脂质过氧化标志物(4-HNE、BodipyC11)在膜破裂前于线粒体富集区域逐渐累积,且可被抗氧化剂 NAC 抑制。该过程体现为线粒体将 ROS 定向输送至细胞周边(该过程命名为mitoxyperiosis),最终导致膜破裂和细胞裂解(该过程命名为mitoxyperilysis)(图3)。
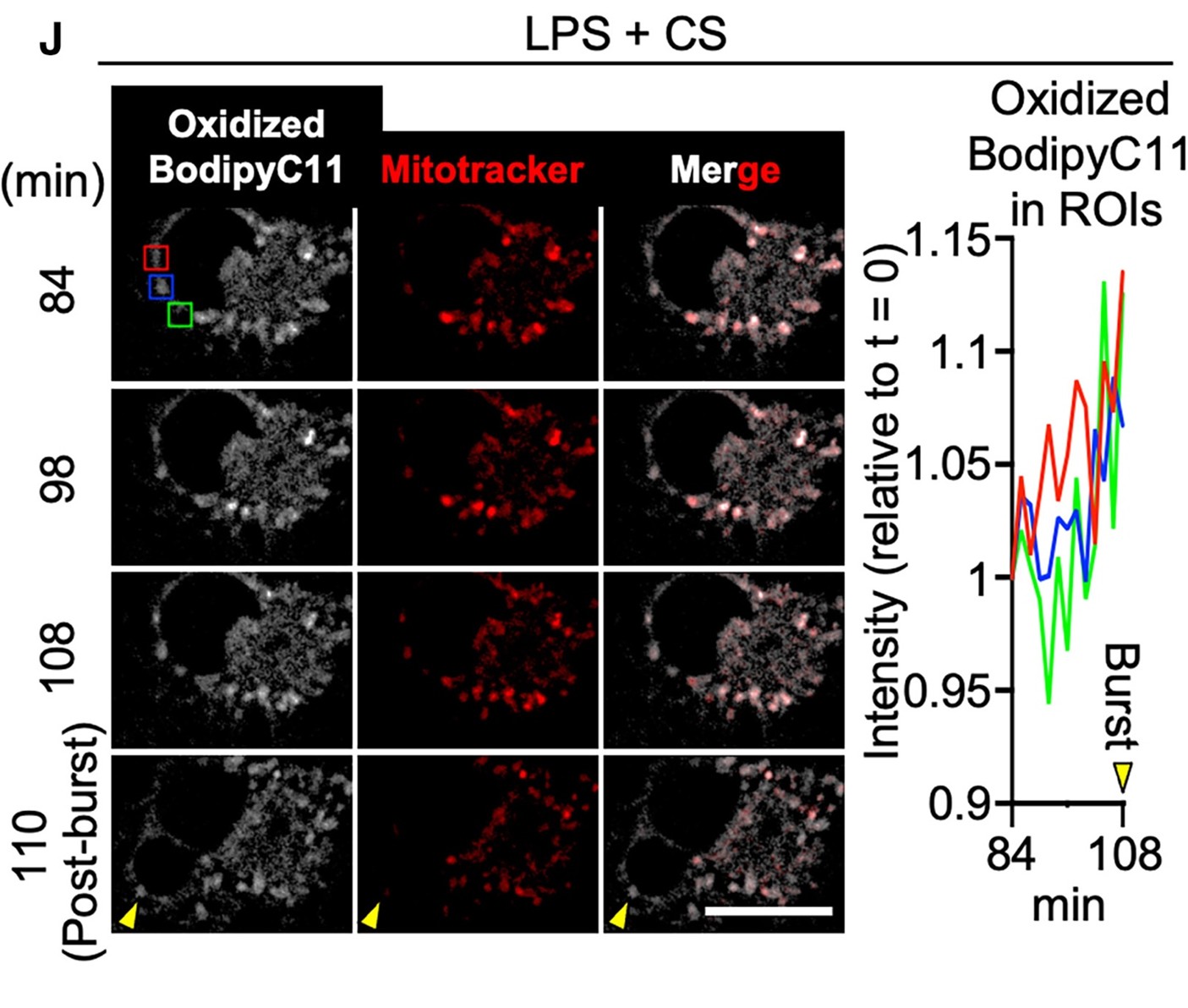
图3. 线粒体锚定在质膜上,通过线粒体过氧化作用诱导局部膜氧化损伤
进一步研究发现IIAMD 通过 BAX/BAK1/BID 诱导线粒体损伤和氧化应激,并激活 mTORC2 信号通路。mTORC2 抑制 RhoA–肌动蛋白介导的伪足形成,降低细胞膜动态,从而延长受损线粒体与质膜的接触,促进局部氧化损伤(mitoxyperiosis)并最终导致膜裂解(mitoxyperilysis)。抑制 mTORC2 可恢复细胞骨架活动,使线粒体远离质膜,在不降低氧化应激的情况下维持膜完整性并阻断细胞死亡(图4)。
最后研究人员证明IIAMD 在体内也是通过 mTORC2 依赖的线粒体–质膜损伤机制(mitoxyperilysis)驱动炎症性组织损伤和肿瘤细胞死亡,既独立于 GSDMD,又可被治疗性调控以抑制肿瘤生长。
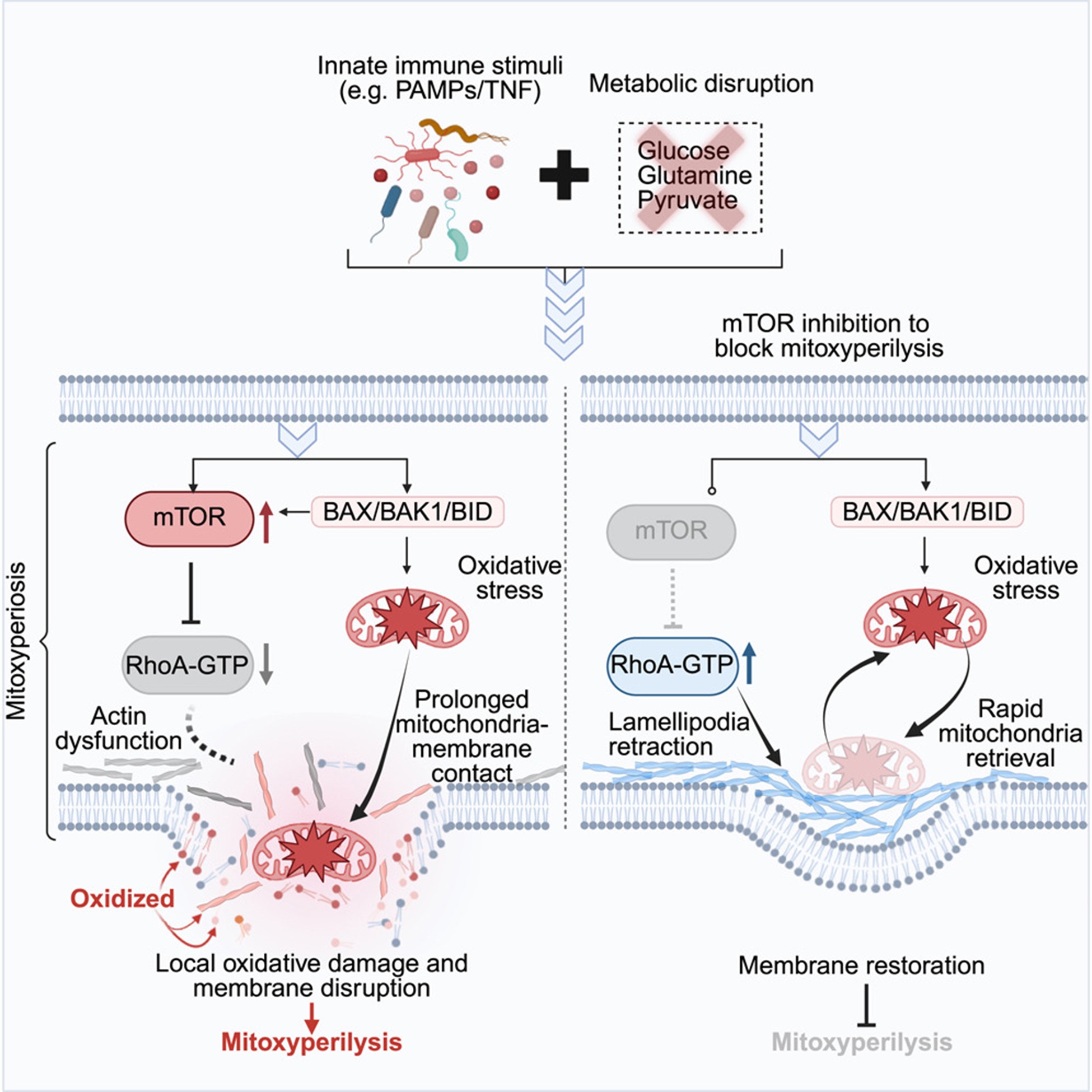
图4. 由先天免疫信号与代谢紊乱协同诱导的全新裂解性细胞死亡模式

