






铁–硫(Fe–S)簇是多种关键细胞过程所必需的古老辅因子,其在线粒体基质中的合成依赖由 NFS1、ISCU2、FDX2 和 frataxin 等组成的多蛋白复合物。Frataxin作为 NFS1 的变构激活因子,其功能缺失会导致弗里德赖希共济失调,但其在 Fe–S 簇生物发生中的精确作用机制仍不清楚。此前研究发现,低氧条件可部分挽救frataxin 缺失引起的 Fe–S 簇不足,为解析其分子机制和发现潜在治疗靶点提供了重要切入点。
来自哈佛医学院Vamsi K. Mootha和Gary Ruvkun团队合作近日在Nature上发表研究,研究利用秀丽隐杆线虫(C.elegans)开展全基因组正向遗传筛选,鉴定出一个能够绕过 frataxin 需求,恢复 Fe–S 簇合成并缓解弗里德赖希共济失调相关表型的抑制突变。

研究人员首先利用了 frataxin/frh-1 空突变秀丽隐杆线虫在低氧条件下可存活的特性,开展正向遗传筛选,鉴定能够使其在非许可性氧条件下恢复生长的抑制突变(图1a)。全基因组测序显示,这些显性抑制突变定位于铁–硫簇组装复合物中的 nfs-1 或 fdx-2 基因,且均为高度保守残基上的错义突变(图1b)。通过 CRISPR 重建实验验证,这些突变可在常氧甚至高氧条件下部分恢复 frataxin 缺失线虫的生长和发育缺陷(图1c)。这一结果表明,调节铁–硫簇组装复合物关键组分的功能可在一定程度上绕过对 frataxin 的依赖。
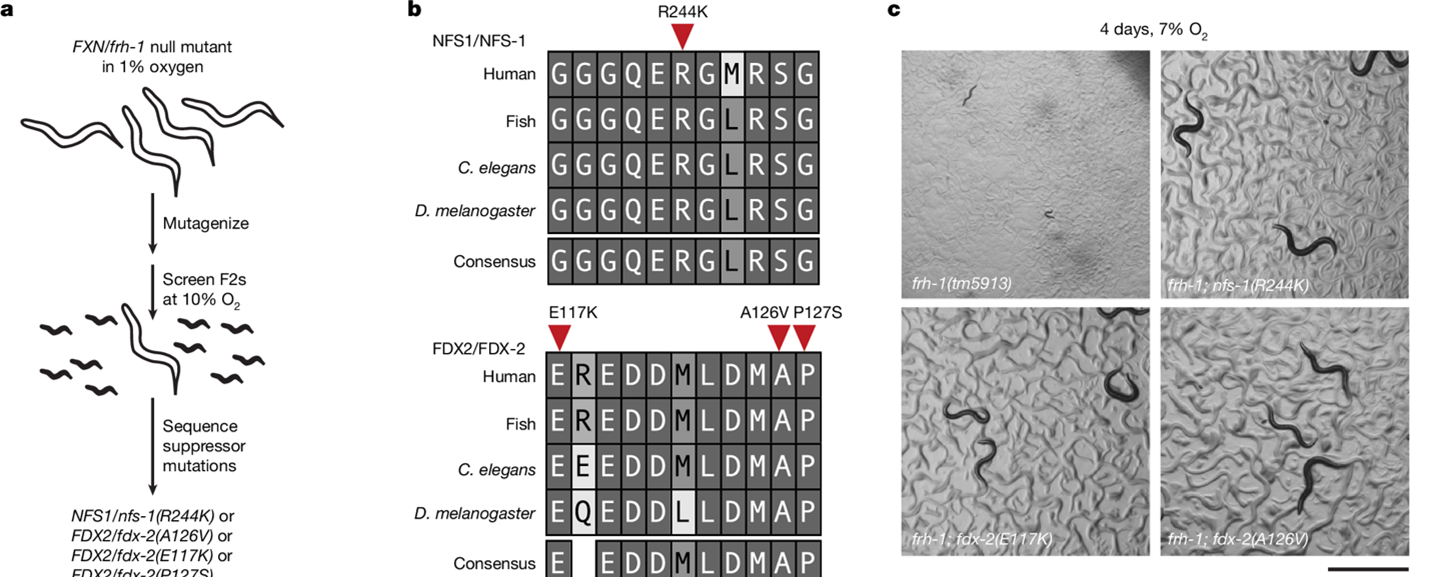
图1. 全基因组正向遗传筛选出位于铁–硫簇组装复合物中的抑制突变
为了解释 nfs-1 和 fdx-2 抑制突变如何挽救 frataxin 空突变动物的生长缺陷,研究人员首先分析了秀丽隐杆线虫中多种应激反应的转录报告基因。他们发现frataxin缺失在线虫中诱导线粒体应激并导致 Fe–S 簇相关蛋白,尤其是电子传递链复合体 I 的显著下降(图2)。fdx-2 和 nfs-1抑制突变可在不全面恢复 Fe–S 簇水平的情况下,部分改善线粒体功能并恢复复合体 I 和 II 的关键亚基。蛋白质组学和生化验证表明,电子传递链通量的恢复是这些突变挽救生长缺陷的核心机制。通过表达可绕过复合体I 和 II 的酵母 NDI1 即可部分恢复 frataxin 突变体的生长,表明增强电子传递链通量是生长挽救的关键机制。
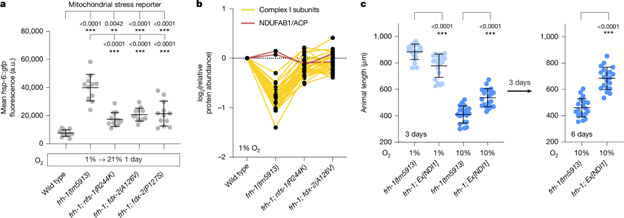
图2. 线粒体铁氧还蛋白FDX2的突变可抑制frataxin缺乏症
遗传互作分析显示,nfs-1(R244K)与 fdx-2(A126V)双突变在野生型背景中导致 Fe–S 簇缺陷相关的生长迟缓和不育,且该表型可被低氧挽救,表明两者通过同一通路作用。结构分析显示,所有抑制突变均位于NFS1–FDX2 相互作用界面,而该界面同时也是 frataxin 的结合位点(图3)。体外生化实验表明,过量 FDX2 会竞争性抑制frataxin 对 NFS1 的激活,从而抑制 Fe–S 簇合成。相反,FDX2(E131K) 等抑制突变不仅减弱 NFS1–FDX2 结合,还可在缺乏frataxin 的情况下增强 Fe–S 簇合成,表现出类似 frataxin 的激活功能。
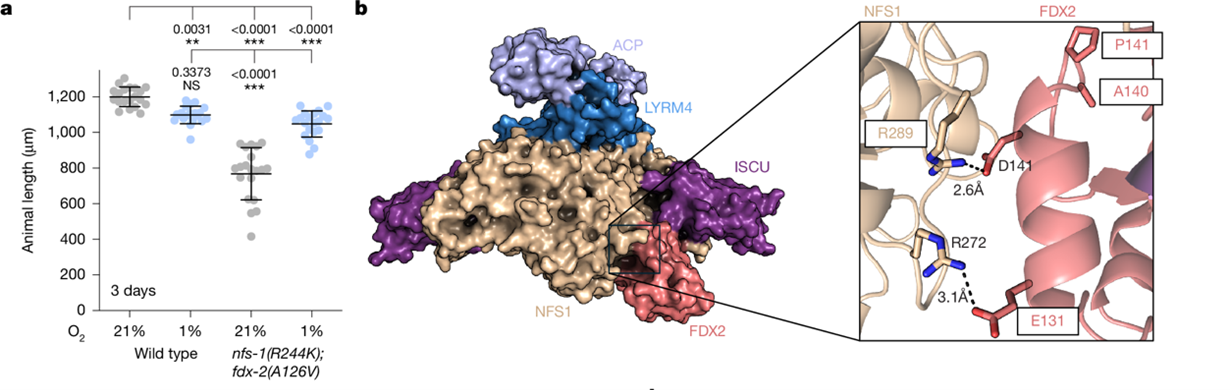
图3. 线粒体铁氧还蛋白FDX2的突变可抑制frataxin缺乏症
研究人员推测,在 frataxin 突变体中,仅仅降低 FDX2 的水平就可能增强 Fe–S 簇的生成。结果显示在线虫中,去除一份 fdx-2 拷贝即可部分挽救 frataxin 缺失导致的生长和发育缺陷(图4),其效果与筛选获得的 fdx-2 抑制突变相当,说明该抑制效应源于单倍体不足性。进一步在人类和小鼠中证实,携带一份 FDX2 功能缺失等位基因是可耐受的。在小鼠 Friedreich 共济失调模型中,Fdx2 杂合突变显著改善了 frataxin 敲低引起的运动障碍。整体结果支持一个模型:在 frataxin 缺失条件下,降低 FDX2 剂量可通过减少其与 NFS1 的抑制性结合而恢复 Fe–S 簇合成并缓解疾病表型。
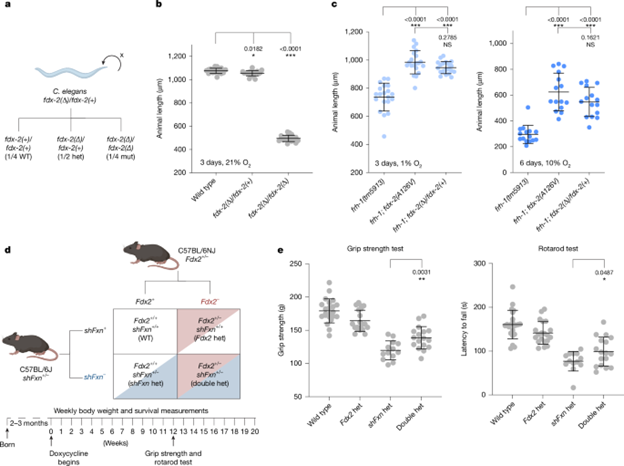
图4. 野生型FDX2活性部分丧失可抑制frataxin突变体

